Bioquímica
HEMOSTASIA Y COAGULACIÓN SANGUÍNEA
 Mecanismo de defensa del organismo con varios cometidos que limitan la pérdida de sangre:
Mecanismo de defensa del organismo con varios cometidos que limitan la pérdida de sangre:
- Mantener permeable la luz vascular.
- Establecer tapón hemostático en caso de lesión vascular.
- Generar la lisis del coágulo de fibrina en caso de obstrucción vascular.
Tipos de Hemostasia
- Hemostasia primaria: Fase vascular y fase plaquetaria.
Hemostasia secundaria: Fase plasmática y fase fibrinolítica.
2) Adhesión plaquetaria:
Al colágeno subendotelial expuesto tras el traumatismo, a través de la glucoproteína de membrana plaquetaria Ib., y mediado por el factor von Willebrand (factor vW) sintetizado en el endotelio.
3) Activación plaquetaria: A medida que las plaquetas se adhieren al endotelio se activan (también lo hacen por la trombina), ocurriendo una serie de hechos trascendentales:
- Cambio de forma. Pasan de discos aplanados a esferas que emiten múltiples pseudópodos, y al mismo tiempo reorganizan el citoesqueleto celular.
- Liberación y oxidación del ácido araquidónico a través de la enzima ciclooxigenasa, para formar finalmente tromboxano A (TxA2), que a su vez induce vasoconstricción y agregación plaquetaria
- Reordenamiento de fosfolipoproteínas de membrana, con capacidad de ligar el factor X y activar la coagulación sanguínea.
- Secreción de gránulos plaquetarios (ADP, PDGF, Serotonina, calcio, etc.), con capacidad de reclutar más plaquetas, aumentar la actividad plaquetaria y reclutar células inflamatorias y fibroblastos para el proceso de reparación.
4) Agregación plaquetaria: Cuando las plaquetas son expuestas a alguno de los agonistas que inician la activación (ADP, TxA2, Trombina, colágeno) comienzan a expresar glicoproteína IIb/IIIa en su superficie, que reconoce dos secuencias presentes en el fibrinógeno y permite formar puentes entre plaquetas activadas.
Hemostasia secundaria o plasmática: coagulación propiamente dicha. Su finalidad es la formación de un coágulo estable de fibrina. Los factores de la coagulación se pueden subdividir en los siguientes grupos:
1) Factores dependientes de la vitamina K: Tienen síntesis hepática, actuando como coenzima la vitamina K, que es necesaria para la carboxilación del ácido glutámico, imprescindible para reaccionar con el calcio y con los fosfolípidos plaquetarios y tisulares.
Son factores dependientes de vitamina K la protrombina o factor II, VII, IX, X y las proteínas C y S.
2) Factores sensibles a la trombina: Fibrinógeno o factor I, y los factores V, VIII y XIII. Además, activa la proteína C.
3) Factores del sistema de contacto (cuando la sangre contacta con una superficie eléctricamente negativa). Constituyen los primeros pasos de la coagulación y son los factores XII, XI, quininógeno de alto peso molecular y precalicreína.
Además de estos factores de coagulación, que son proteínas plasmáticas, son necesarios fosfolípidos de las plaquetas y los tejidos, y calcio, que actúa como puente entre ambos grupos.
Cascada de coagulación
Vías de la coagulación

Tras las convergencias de ambas vías en los factores X y V se produce posteriormente la activación de la protrombina o factor II en trombina, que a su vez dará lugar a:
• Formación de fibrina a partir de fibrinógeno o factor I.
• Agregación plaquetaria y secreción de gránulos plaquetarios (es decir, la trombina produce una nueva reactivación de la hemostasia primaria).
• Activación de los factores V, VIII, XI y XIII.
• Activación de la proteína C.
• Activación del inhibidor de la fibrinolisis activado por trombina (TAFI).
FACTORES DE COAGULACIÓN
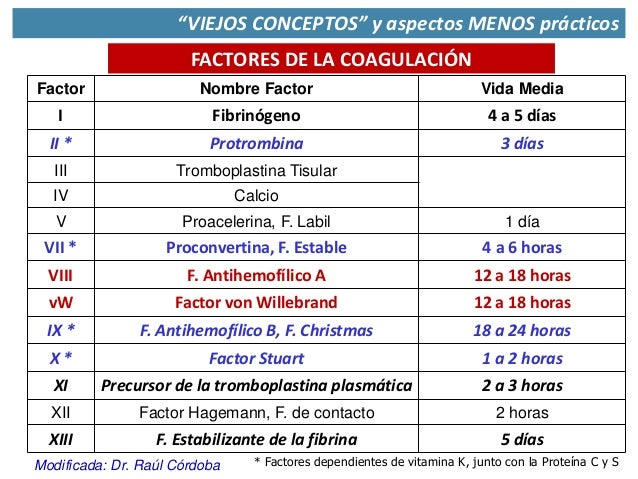
•Factores XII, XI, X, VII, II y la precalicreína (PK) son zimógenos de proteasas de serina.
•Factores V y VIII y HMWK (kininógeno de alto peso molecular) son cofactores y deben activarse para funcionar.
•FT (factor tisular de membrana celular) se activa al hacer contacto con la sangre.
•Factores II, VII, IX y X dependen de la vitamina K para su síntesis hepática.
OTROS FACTORES
•Prekalikreina (factor Fletcher)
•Kininógeno (HMWK, factor Fitzgerald)
•Factor Von Willebrand
•Proteína C
•Proteína S
•Antitrombina III
•El FT es el único factor que no se encuentra normalmente en la circulación sanguínea, es una proteína específica presente sobre la membrana plasmática de células como monocitos o células endoteliales. El FT se activa únicamente al entrar en contacto con el FVII, momento en el que se inicia la coagulación plasmática.
•La TM se expresa sobre las células del endotelio vascular, participa como anticoagulante activando a la proteína C.
Alteraciones de la Coagulación y Factores Relacionados
|
Factor |
Enfermedad |
|
Fibrinógeno |
Afibrinogenemia,
hipofibrinogenemia |
|
Protrombina |
Hipoprotrombinemia |
|
Factor V |
Parahemofilia |
|
Factor VII |
Déficit congénito del factor
VII |
|
Factor VIII |
Hemofilia A |
|
Factor IX |
Hemofilia B |
|
Factor XI |
Hemofilia C |
|
Factor XII |
Síndrome de Hageman |
|
Precalicreína |
Sindrome de Fletcher |
|
Quininógeno de alto peso
molecular (HMWK) |
Síndrome de Fitzgerald |
|
Factor Von Willebrand |
Enfermedad de Von Willebrand |


Comentarios
Publicar un comentario